
Resumen
Nuestro grupo posee como interés principal dilucidar las bases genéticas de enfermedades humanas.
Aplicamos diferentes metodologías y diferentes abordajes para hallar variaciones en el material genético que nos orienten sobre la contribución en la causalidad de algunas enfermedades hereditarias, así como de otras enfermedades en donde el componente genético influye en combinación con otros factores como pueden ser factores ambientales, dietarios, etc.
Estamos enfocados tanto en el análisis de genes individuales como en el estudio de a nivel genómico. Con esta información, y para algunos genes de interés, profundizamos en los posibles efectos biológicos que poseen estas variaciones. Los abordajes incluyen análisis bioinformáticos predictivos así como ensayos de laboratorio que nos permitan determinar la funcionalidad de los cambios observados.
Las enfermedades humanas en las cuales estamos trabajando en la actualidad son: Deficiencia de 21-hidroxilasa; Insuficiencia Ovárica Primaria; Cardiopatías congénitas
Líneas de Investigación
El déficit de 21-hidroxilasa comprende un espectro que abarca formas severas con pérdida salina al nacimiento, hasta aquellas con signos leves de hiperandrogenismo fuera del período neonatal. El gen codificante, CYP21A2, se localiza en el cromosoma 6 en una región de alta densidad génica llamada módulo RCCX, cuya estructura comprende un arreglo en tándem y duplicado conteniendo genes activos y pseudogenes. La mayor parte de las causas genéticas de la deficiencia se produce por rearreglos genómicos en esta región. Nuestro grupo de trabajo investiga esta patología desde el año 1996 y contribuyó con una caracterización molecular de más de 1500 individuos de nuestra población, el reporte de variantes noveles así como de sus efectos biológicos. Actualmente, estamos abocados a analizar esta región mediante secuenciación masiva de 3era generación que nos permite profundizar en todos los rearreglos genómicos que presenta esta región.
Las CC abarcan un amplio espectro de entidades que poseen anomalías en la estructura del corazón y los grandes vasos. Poseen una prevalencia mundial entre 6-9/1.000 nacimientos y se destacan como causa de morbi-mortalidad prenatal, neonatal e infantil. La etiología es mayoritariamente multifactorial, en donde intervendrían factores ambientales y genéticos. Los factores genéticos abarcan desde anomalías cromosómicas hasta variantes de secuencia patogénicas en un solo gen. Nuestro grupo de trabajo inició en el año 2013 esta línea de investigación con el objetivo de identificar diferentes defectos genéticos en afectados con CC en nuestra población. Actualmente, las investigaciones están centradas en los estudios masivos del genoma para identificar los genes involucrados, y en realizar análisis predictivos mediante abordajes bioinformáticos y funcionales de variantes de secuencia en el gen NKX2-5.
El gen FMR1 está implicado, por diferentes mecanismos moleculares, en 4 patologías de origen genético, entre ellas, la Insuficiencia Ovárica Primaria Asociada a la Fragilidad del X (FXPOI). En nuestro grupo estudiamos la expresión del gen en un modelo de foliculogénesis en rata y la posible regulación del mismo por microRNAs. Nos interesa también caracterizar las isoformas de splicing que se expresan en el tejido ovárico en distintos estadíos del desarrollo folicular y en muestras de células de granulosa humanas, para lo cual realizamos secuenciaciones de 3ra generación que permiten una mejor caracterización de las isoformas.
Galeria:

Dr. Alejandro Nadra, Dra Liliana Dain, Dr. Javier Santos 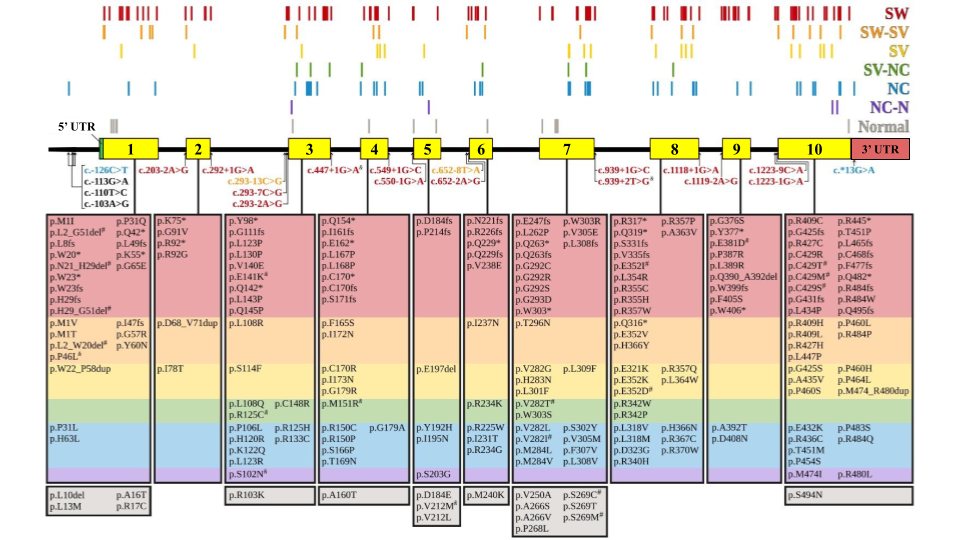
Genética molecular de la deficiencia de 21-hidroxilasa 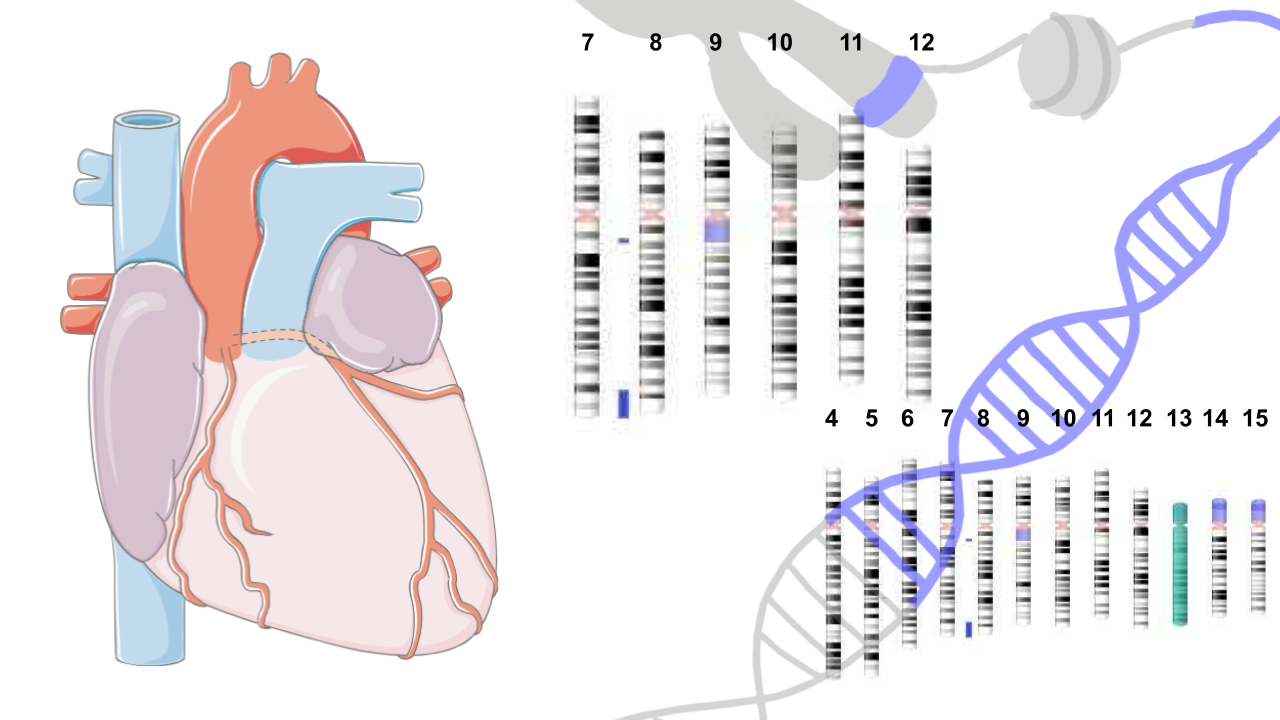
Causas genéticas de las cardiopatías congénitas (CC) 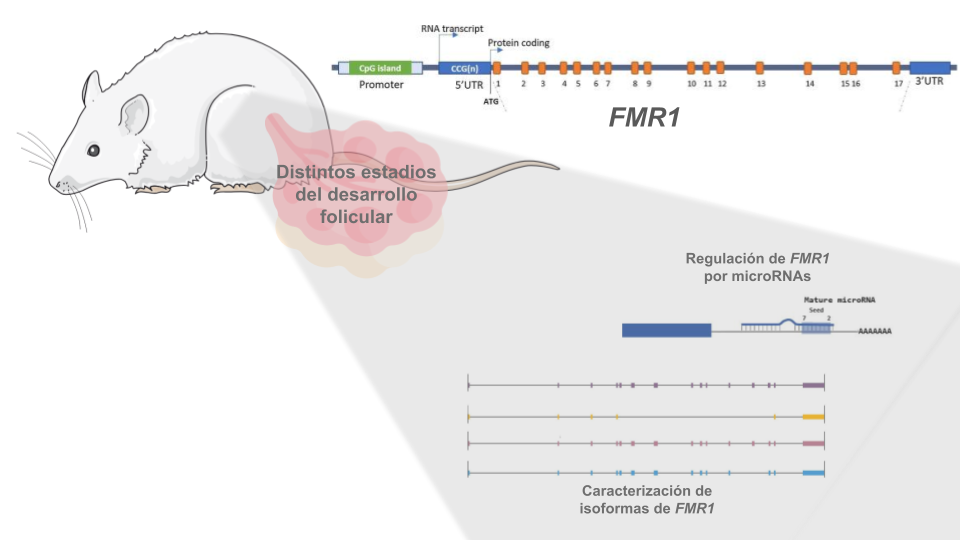
Rol del gen FMR1 en la función ovárica
Integrantes:
Ianina Ferder
Investigadora asistente CONICET
iferder@fbmc.fcen.uba.ar
Melisa Taboas
meivta84@gmail.com
Profesional principal
CNGM-ANLIS
Aldana Claps
Profesional adjunto
CNGM-ANLIS
Julieta Laiseca
Profesional adjunto
CNGM-ANLIS
Emilio Kolomenski
Becario doctoral CONICET
ekolomenski@gmail.com
Yamila Gandola
Becaria post-doctoral
AMPCyT
yamigandola@gmail.com
Marina Luz Ingravidi
Becaria doctoral CONICET
maru.luz98@hotmail.com
Colaboradores:
Dra Laura Kamenetzky
iB3, FCEN, UBA
Dr. Alejandro Nadra
iB3, FCEN, UBA
Dr. Javier Santos
iB3, FCEN, UBA
Publicaciones destacadas:
- Ferder IC, Espeche LD, Bruque CD, Parborell F, Tesone M, Dain L, 2022
Expression and characterisation of Fmr1 splice variants during folliculogenesis in the rat
Reproduction, Fertility and Development, 34(16):1034-1042
10.1071/RD22059 - Delea M, Massara LS, Espeche LD, Bidondo MP, Barbero P, Oliveri J, Brun P, Fabro
M, Galain M, Fernández CS, Taboas M, Bruque CD, Kolomenski JE, Izquierdo A,
Berenstein A, Cosentino V, Martinoli C, Vilas M, Rittler M, Mendez R, Furforo L,
Liascovich R, Groisman B, Rozental S, Dain L, 2022
Genetic Analysis Algorithm for the Study of Patients with Multiple Congenital Anomalies and Isolated Congenital Heart Disease
Genes (Basel). 29;13(7):1172
10.3390/genes13071172 - Espeche LD, Sewell KE, Castro IH, Capece L, Pignataro MF, Dain L, Santos J., 2022
Conformational stability, dynamics and function of human frataxin: Tryptophan side
chain interplay
Archives of Biochemistry and Biophysics, 15;715:109086
10.1016/j.abb.2021.109086 - Kolomenski JE, Delea M, Simonetti L, Fabbro MC, Espeche LD, Taboas M, Nadra AD, Bruque CD, Dain L., 2020
An update on Genetic Variants of the NKX2-5
Human Mutation, 41(7):1187-1208
10.1002/humu.24030 - Fernández CS, Taboas M, Bruque CD, Benavides-Mori B, Belli S, Stivel M, Oneto A,
Pasqualini T, Delea M, Espeche LD, Kolomenski JE, Alba L, Buzzalino N, Dain L, 2020
Genetic Characterization of a Large Cohort of Argentine 21-Hydroxylase Deficiency
Clinical Endocrinology (oxf), 93(1):19-27
10.1111/cen.14190 - Simonetti L, Bruque CD, Fernández CS, Benavides-Mori B, Delea M, Kolomenski JE,
Espeche LD, Buzzalino ND, Nadra AD, Dain L., 2018
CYP21A2 mutation update: Comprehensive analysis of databases and published genetic variants
Human Mutation, 39:5–22
10.1002/humu.23351 - Espeche LD, Chiauzzi V, Ferder I, Solari AP, Bruque CD, Delea M, Belli S, Fernández
CS, Buzzalino ND, Charreau E, Dain L, 2017
Distribution of FMR1 and FMR2 repeats in Argentinean patients with primary ovarian insufficiency
Genes, 16;8(8). pii: E194
10.3390/genes8080194 - Bruque CD, Delea M, Fernández CS, Orza JV, Taboas M, Buzzalino N, Espeche LD, Solari A, Luccerini V, Alba L, Nadra AD, Dain L, 2016
Structure-based activity prediction of CYP21A2 stability variants: A survey of available gene variations
Scientific Reports, 6, 39082
10.1038/srep39082 - Taboas M, Gómez Acuña L, Scaia MF, Bruque CD, Buzzalino N, Stivel M, Ceballos
NR, Dain L., 2014
Functional Studies of p.R132C, p.R149C, p.M283V, p.E431K, and a Novel c.652-2A.G Mutations of the CYP21A2 Gene
PLoS ONE, 9(3): e92181
10.1371/journal.pone.0092181
Prensa:
Nexciencia , 2022
INVESTIGACIÓN EN RECIÉN NACIDOS: GENES ALTERADOS
https://nexciencia.exactas.uba.ar/desarrollan-algoritmo-identificar-causas-geneticas-anomalias-bebes-en-argentina-liliana-dain
Nexciencia , 2019
ESTUDIOS EN INFANTES: PROBLEMAS DEL CORAZÓN ARGENTINO
https://nexciencia.exactas.uba.ar/problemas-trastornos-corazon-congenitos-sindrome-microdelecion-22q11-genetica-liliana-dain-marisol-delea
Agencia CyTA, 2018
CREAN BASE DE DATOS PARA FAVORECER EL DIAGNÓSTICO DE UNA RARA ENFERMEDAD CONGÉNITA
https://www.agenciacyta.org.ar/2018/05/crean-base-de-datos-para-favorecer-el-diagnostico-de-una-rara-enfermedad-congenita/
